
VYJUVEK 5 x 1 000 000 000 unités formant plages -mL, de suspension et gel pour gel, boîte de 1 flacon de 2,50 ml
Dernière révision : 26/06/2025
Taux de TVA : 0%
Laboratoire exploitant : PHARMA BLUE
Source :

Vyjuvek est indiqué pour le traitement des plaies chez les patients atteints d'épidermolyse bulleuse dystrophique (EBD) présentant une ou plusieurs mutations du gène de la chaîne alpha 1 du collagène de type VII (COL7A1), dès la naissance.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité du médicament biologique, le nom et le numéro de lot du médicament administré doivent être clairement enregistrés.
Carcinome épidermoïde
Vyjuvek ne doit pas être appliqué sur des plaies présentant un diagnostic confirmé ou suspecté de carcinome épidermoïde. Vyjuvek peut tout de même être appliqué sur les autres plaies chez les patients qui développent un carcinome épidermoïde.
Transmission d'un agent infectieux
Bérémagène géperpavec ne se réplique pas dans les cellules. Il ne s'intègre pas à l'ADN natif et n'interagit pas avec celui-ci.
Bien que bérémagène géperpavec soit stérile, il existe un risque de transmission d'agents infectieux. Les professionnels de santé qui administrent Vyjuvek doivent par conséquent surveiller les patients afin de détecter tout signe ou symptôme d'infection après le traitement et de les traiter de façon adéquate, si nécessaire.
Les personnes qui manipulent bérémagène géperpavec ou qui aident à changer les pansements doivent porter un équipement de protection (voir la rubrique Précautions particulières d’élimination et de manipulation).
Les femmes enceintes ne doivent pas manipuler les déchets relatifs aux pansements. Les aidants ou les professionnels de santé qui appliquent le gel doivent respecter l'obligation de recouvrir les plaies par des pansements. Il convient également de conseiller aux patients d'éviter de toucher ou de gratter les plaies afin d'empêcher toute contamination sur d'autres parties du corps ou de son entourage.
Suivi à long terme
Il est recommandé que les patients participent à une étude non interventionnelle internationale afin d'évaluer la sécurité à long terme de bérémagène géperpavec en conditions de vie réelle.
Résumé du profil de sécurité
Dix-huit patients (58 %) ont signalé au moins un effet indésirable lors de l'essai clinique. Les effets indésirables les plus couramment signalés étaient les frissons (9,7 %) et le prurit (9,7 %).
Aucun effet indésirable n'a conduit à l'arrêt du traitement.
Tableau des effets indésirables
Sauf indication contraire, les fréquences des effets indésirables sont basées sur toutes les fréquences des événements indésirables identifiés chez 31 patients exposés à bérémagène géperpavec pendant une durée médiane de 25 semaines dans l'étude de phase 3 randomisée et contrôlée par placebo chez un même sujet. Voir la rubrique Propriétés pharmacodynamiques pour des informations sur les principales caractéristiques des patients inclus dans l'essai clinique.
Dans le tableau suivant, les effets indésirables sont répertoriés par classe de systèmes d'organes selon MedDRA, par terme privilégié et par fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
La fréquence des effets indésirables est définie comme suit: très fréquent (≥ 1/10); fréquent (≥ 1/100 à < 1/10); peu fréquent (≥ 1/1 000 à < 1/100); rare (≥ 1/10 000 à < 1/1 000); très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3. Effets indésirables
Population pédiatrique
Sur les 31 sujets de l'étude de phase 3, 19 (61 %) étaient des sujets pédiatriques (âgés de 17 ans ou moins), dont 3 (9,7 %) âgés de 3 ans ou moins. Sur les 19 patients pédiatriques, 8 étaient de sexe féminin (42 %).
Compte tenu de l'identité du produit, de son mode d'administration et de son confinement localisé, la fréquence, le type et la gravité des effets indésirables chez les enfants devraient être les mêmes que chez les adultes.
Immunogénicité
Les preuves d'une exposition systémique au vecteur viral après une application cutanée de Vyjuvek sont limitées. Les anticorps dirigés contre le vecteur (HSV-1) et la protéine codée par le transgène (COL7) ont été évalués dans un sous-ensemble de participants de l'étude clinique randomisée et contrôlée par placebo chez un même sujet. Au total, 64 % des sujets évalués (14/22) étaient positifs aux anticorps anti-HSV-1 au début de l'étude. Six des huit sujets séronégatifs anti-HSV-1 ont été séroconvertis au cours de la 26e semaine après un traitement par Vyjuvek. Chez les sujets pour lesquels des échantillons de sérum de référence et de fin d'étude correspondants étaient disponibles, des anticorps antimédicaments (ADA) dirigés contre COL7 ont été détectés chez 72 % (13/18) des sujets traités par Vyjuvek pendant 26 semaines au maximum. Aucune réponse immunitaire neutralisant n'a été observée lors d'une exposition initiale ou répétée à Vyjuvek. L'incidence de la séroconversion sur le maintien de l'effet du traitement est inconnue, car les données ne sont pas disponibles après 26 semaines.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
PRECAUTIONS à prendre :
- ÉVITER tout contact direct avec les plaies traitées (par exemple,
toucher ou gratter) et les pansements des plaies traitées pendant
environ 24 heures après le traitement.
- PORTER un équipement de protection individuelle (gants, masque,
protection des yeux, par exemple) lors de l'administration du produit,
du changement des pansements et de leur élimination.
- En cas d'exposition accidentelle (par exemple, par une éclaboussure
dans les yeux ou les muqueuses), RINCER à l'eau pendant au moins 5
minutes.
- En cas d'exposition d'une peau intacte, NETTOYER soigneusement la
zone affectée avec du savon et de l'eau et/ou un désinfectant.
- Femme enceinte : NE PAS
préparer ou administrer ce médicament, EVITER tout contact direct
avec les plaies traitées ou avec les pansements des plaies traitées.
Grossesse
Il n'existe pas de données sur l'utilisation de bérémagène géperpavec chez la femme enceinte. Les études effectuées chez l'animal sont insuffisantes pour tirer des conclusions sur la toxicité pour la reproduction (voir la rubrique Données de sécurité préclinique).
L'utilisation de Vyjuvek n'est pas recommandée pendant la grossesse.
Allaitement
On ne sait pas si bérémagène géperpavec est excrété dans le lait maternel.
Un risque pour les nouveau-nés/nourrissons ne peut pas être exclu.
Une décision doit être prise soit d'interrompre l'allaitement, soit d'interrompre le traitement par Vyjuvek ou de s'en abstenir en tenant compte du bénéfice de l'allaitement maternel pour l'enfant et du bénéfice du traitement pour la femme.
Fertilité
Aucun essai non-clinique ni aucune étude clinique n'ont été réalisés pour évaluer l'effet de bérémagène géperpavec sur la fertilité.
Aucune étude d'interaction n'a été réalisée avec Vyjuvek. Les interactions avec des médicaments topiques n'ont pas été étudiées dans le cadre d'essais cliniques. Les autres médicaments topiques ne doivent pas être administrés en même temps que Vyjuvek.
La sécurité de l'immunisation par les vaccins viraux vivants pendant ou après le traitement par Vyjuvek n'a pas été étudiée. Il n'existe aucune donnée suggérant que Vyjuvek pourrait avoir une incidence sur la capacité de l'organisme à réagir de manière appropriée à un vaccin contre le virus vivant.
Le traitement par Vyjuvek doit être initié par des professionnels de santé expérimentés dans la prise en charge des patients atteints d'épidermolyse bulleuse dystrophique.
Posologie
Vyjuvek est appliqué par voie cutanée sur une ou plusieurs plaies, une fois par semaine, en petites gouttelettes réparties sous forme de quadrillage, espacées l'une de l'autre d'environ 1 cm. Il est possible que toutes les plaies ne puissent pas être traitées à chaque séance de traitement.
La dose hebdomadaire totale maximale recommandée pour les enfants de la naissance à l'âge de 3 ans est de 1 mL (2×109 UFP). La dose totale hebdomadaire maximale recommandée pour les enfants âgés de plus de 3 ans, les adolescents et les adultes est de 2 mL (4×109 UFP).
Vyjuvek doit être appliqué sur les plaies jusqu'à ce qu'elles soient refermées avant de sélectionner une ou plusieurs nouvelles plaies à traiter. Le traitement hebdomadaire des plaies précédemment traitées doit être privilégié en cas de réouverture. Vyjuvek ne doit pas être appliqué en l'absence de plaies.
Le tableau ci-dessous sert de référence pour la dose en fonction de la taille approximative de la plaie chez les enfants, les adolescents et les adultes.
Tableau 1. Dose par surface de la plaie
|
Surface de la plaie (cm2)* |
Dose (UFP)a |
Volume (mL) |
|
< 20 |
< 4×108 |
< 0,2 |
|
20 à < 40 |
4×108 à 8×108 |
0,2 à < 0,4 |
|
40 à 60 |
8×108 à < 1,2×109 |
0,4 à < 0,6 |
|
60 à < 200 |
1,2×109 à < 4×109 |
0,6 à < 2 |
UFP = unités formant plage a: La dose maximale chez les enfants de moins de 3 ans est de 1 mL (2×109) UFP.
En cas d'oubli d'une dose, Vyjuvek doit être administré dès que possible et la posologie hebdomadaire doit ensuite être reprise.
Populations particulières
Sujets âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés de 65 ans ou plus.
Mode d'administration
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient des organismes génétiquement modifiés (voir la rubrique Mises en garde spéciales et précautions d'emploi). Au cours de la préparation, de l'application et de l'élimination, des précautions appropriées doivent être prises. Un équipement de protection individuelle (p. ex. des gants, un masque et une protection oculaire) doit être porté lors de la manipulation de Vyjuvek.
Les femmes enceintes ne doivent pas préparer ou administrer Vyjuvek et doivent éviter tout contact direct avec les plaies traitées ou avec les pansements des plaies traitées (voir la rubrique Précautions particulières d'élimination et de manipulation).
Administration
Pour usage cutané uniquement sur des plaies.
Avant toute utilisation cutanée, la suspension et le gel doivent être décongelés, et la suspension doit être mélangée au gel dans une pharmacie hospitalière. Pour des instructions détaillées sur la préparation, la durée de conservation après mélange, l'administration, les mesures à prendre en cas d'exposition accidentelle, la logistique et l'élimination de Vyjuvek, voir les rubriques Durée de conservation et Précautions particulières d'élimination et de manipulation.
Un professionnel de santé doit appliquer Vyjuvek, dans un établissement de santé (p. ex. en milieu hospitalier) ou à domicile. Si le professionnel de santé le juge approprié, des patients ou des aidants formés peuvent également appliquer Vyjuvek.
Les plaies doivent être nettoyées délicatement à l'aide d'un produit ne contenant pas d'agent virucide avant l'application cutanée. Les médicaments et les pommades appliqués sur la plaie doivent être éliminés et celle-ci doit être nettoyée avant l'administration de Vyjuvek afin de ne pas diminuer son activité (voir la rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Tableau 2. Étapes de l'administration
|
Étape 1. La seringue contenant Vyjuvek doit être amorcée avant la première application en tirant le piston vers le bas et en le poussant vers le haut, de sorte qu'une gouttelette de Vyjuvek se forme à l'extrémité de la seringue. |
|
|
Étape 2. Vyjuvek doit être appliqué sur la plaie sélectionnée, en petites gouttelettes espacées d'environ 1 cm (largeur du bout du doigt), seule la gouttelette doit toucher la plaie. Seul le gel doit entrer en contact avec la peau. L'extrémité de la seringue ne doit pas toucher la peau afin d'éviter une contamination du gel contenu dans la seringue. |
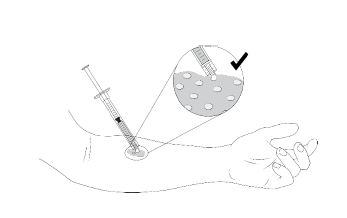
|
|
Étape 3. Une fois que Vyjuvek a été appliqué sur la plaie, un pansement hydrophobe doit recouvrir la plaie. Le pansement doit être coupé à une taille légèrement supérieure à celle de la plaie, mais peut varier en fonction des préférences du patient. Une fois que les gouttelettes de Vyjuvek sont recouvertes par le pansement hydrophobe, une couche mince et uniforme de Vyjuvek se formera sur la plaie. |
|
|
Étape 4. Le pansement standard doit être découpé à une taille plus grande que le pansement hydrophobe. Le pansement standard sera placé sur le pansement hydrophobe pour empêcher le contact du gel avec d'autres zones du corps ou des personnes de l'entourage. |
|
Le pansement doit être laissé en place pendant environ 24 heures après l'application de Vyjuvek. Une fois les pansements de Vyjuvek retirés, le patient peut poursuivre ses soins classiques.
Vyjuvek doit continuer à être administré une fois par semaine jusqu'à la cicatrisation complète des plaies. En cas de réouverture de plaies préalablement traitées, Vyjuvek doit être de nouveau appliqué. Vyjuvek ne doit pas être appliqué en l'absence de plaies.
Durée de conservation :
Cartons non ouverts
2 ans lorsqu'ils sont conservés dans le congélateur.
Après décongélation
Si un congélateur n'est pas disponible, le ou les cartons peuvent être entreposés dans un réfrigérateur (de 2 °C à 8 °C) pendant une durée maximale d'un mois.
Une fois conservé au réfrigérateur, le médicament ne doit pas être recongelé.
Après reconstitution
La stabilité chimique et physique en cours d'utilisation a été démontrée pendant 168 heures (7 jours) à une température comprise entre 2 °C et 8 °C.
D'un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, la durée et les conditions de conservation relèvent de la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures entre 2 et 8° C, sauf si le mélange a eu lieu dans des conditions d'asepsie contrôlées et validées.
Les seringues peuvent être conservées à température ambiante pour une durée maximale de 8 heures.
Conditions de transport d'un produit reconstitué
Transportez le produit reconstitué à une température comprise entre 2 °C et 8 °C jusqu'au lieu d'administration.
Précautions particulières de conservation :
Cartons non ouverts
À conserver congelés entre -15 °C et -25 °C. À transporter congelés (< -20 °C).
Conservez les flacons dans le carton avant la décongélation afin de les protéger de la lumière.
Après décongélation et reconstitution
Pour les conditions de conservation après décongélation et après reconstitution du médicament, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Aucun cas de surdosage de Vyjuvek n'a été signalé. Un traitement symptomatique et de soutien, jugé nécessaire par le professionnel de santé traitant, est conseillé en cas de surdosage.
Classe pharmacothérapeutique: Préparations pour le traitement des plaies et des ulcères, cicatrisants, code ATC: D03AX16
Mécanisme d'action
Bérémagène géperpavec est une thérapie génique basée sur un virus de l'herpès simplex de type 1 (HSV-1) modifié et incompétent pour la réplication et contenant le gène codant pour la protéine COL7A1, ciblant la cause génétique sous-jacente de l'épidermolyse bulleuse dystrophique. Le vecteur HSV-1 appartient à la famille des virus de l'herpès humain (HHV) à ADN double brin. Lors de l'application cutanée sur les plaies, bérémagène géperpavec peut transduire à la fois les kératinocytes et les fibroblastes. Après l'entrée de bérémagène géperpavec dans les cellules, le génome du vecteur est déposé dans le noyau sans s'intégrer dans l'ADN de la cellule hôte et sans autre perturbation. Une fois dans le noyau, la transcription de COL7A1 humain codé par le vecteur est initiée. Les produits de la transcription qui en résultent permettent la production et la sécrétion de COL7 par la cellule dans sa forme mature. Ces molécules de COL7 s'organisent en faisceaux longs et fins pour former des fibrilles d'ancrage. Les fibrilles d'ancrage maintiennent l'épiderme et le derme ensemble et sont essentielles au maintien de l'intégrité de la peau Efficacité et sécurité cliniques
L'efficacité de Vyjuvek chez les sujets âgés d'un an et plus atteints d'EBD, présentant une ou plusieurs mutations du gène COL7A1, a été évaluée dans un essai contrôlé randomisé. Tous les sujets de l'étude présentaient une EBD avec mutation(s) génétiquement confirmée(s) du gène COL7A1. Deux plaies comparables chez chaque sujet ont été sélectionnées et randomisées pour recevoir soit une application cutanée hebdomadaire de bérémagène géperpavec soit un placebo (gel seul) pendant 26 semaines. La dose hebdomadaire maximale totale a été définie en fonction de la tranche d'âge: les sujets âgés de ≥ 6 mois à < 3 ans ont reçu 1,6×109 UFP/semaine, les sujets de ≥ 3 ans à < 6 ans ont reçu 2,4×109 UFP/semaine, et les sujets âgés de ≥ 6 ans ont reçu 3,2×109 UFP/semaine.
L'étude a inclus 31 sujets (20 hommes et 11 femmes), dont 30 sujets atteints d'EBD autosomique récessive et un sujet avec une EBD autosomique dominante. La surface des plaies primaires traitées par bérémagène géperpavec était comprise entre 2 et 57 cm2, 74 % des plaies étant d'une surface < 20 cm2 et 19 % entre 20 et < 40 cm2. La surface des plaies traitées par un gel placebo variait de 2 à 52 cm2, 71 % des plaies étant < 20 cm2 et 26 %, de 20 à < 40 cm2. La plaie secondaire de plus grande taille traitée était ≥ 130 cm2. L'âge moyen des sujets était de 17 ans (de un an à 44 ans), incluant 61 % de sujets pédiatriques (n = 19, âgés de un an à moins de 17 ans) et 9,7 % de sujets ayant moins de trois ans. Soixante-quatre pour cent des sujets étaient blancs ; 19 % étaient asiatiques et les autres étaient des indiens d'Amérique ou originaires d'Alaska.
L'efficacité a été évaluée sur la base d'une amélioration de la cicatrisation des plaies définie comme la différence dans le pourcentage de fermeture complète (100 %) de la plaie à 24 semaines, confirmée lors de deux visites consécutives de contrôle espacées de deux semaines d'intervalle, évaluée aux semaines 22 et 24 ou aux semaines 24 et 26, entre des plaies traitées par bérémagène géperpavec et celles traitées par le gel placebo. L'efficacité a également été évaluée par la différence de pourcentage de cicatrisation complète de la plaie évaluée aux semaines 8 et 10 ou aux deux semaines 10 et 12 entre des plaies traitées par bérémagène géperpavec et celles traitées par le gel placebo. La cicatrisation complète des plaies a été définie comme une fermeture à 100 % de la surface exacte de la plaie sélectionnée à l'inclusion de l'étude, décrite comme une ré-épithélialisation de la peau sans drainage, évaluée lors de deux visites consécutives à deux semaines d'intervalle. Les résultats en matière d'efficacité sont résumés dans le tableau 4.
Tableau 4. Critère d'évaluation principal et critère d'évaluation secondaire clé *
|
Délais d'évaluation de la fermeture de plaie |
Plaies primaires exposées à bérémagène géperpavec (N = 31) |
Plaies primaires exposées au placebo (N = 31) |
Différence absolue (CI à 95 %) |
Valeur p |
|
Critère d'évaluation principal: cicatrisation complète de la plaie après 6 mois†‡ |
20,9 (67 %) |
6,7 (22 %) |
46 (24-68 %) |
0,002 |
|
Critère d'évaluation secondaire clé: cicatrisation complète des plaies après 3 mois‡ |
21,9 (71 %) |
6,1 (20 %) |
51 (29-73 %) |
< 0,001 |
*Les critères d'évaluation principal et secondaire ont été analysés dans la population en intention de traiter. Des méthodes d'imputation multiple ont été utilisées pour tenir compte des données manquantes. Les comptages fractionnés sont dus à la procédure d'imputation multiple utilisée pour l'analyse. Le test d'hypothèse a été réalisé en utilisant le test exact de McNemar.
†Les plaies primaires ont été évaluées aux semaines 22 et 24 ou aux semaines 24 et 26.
‡Les plaies primaires ont été évaluées aux semaines 8 et 10 ou aux semaines 10 et 12.
Dans l'essai de confirmation, des évaluations de l'exposition systémique ont été réalisées lors de visites hebdomadaires sur site clinique par quantification des génomes de bérémagène géperpavec présents dans des échantillons de sang et d'urine (excrétion du vecteur) par un test qPCR validé. Tous les échantillons de sang et tous les échantillons d'urine, sauf un, prélevés tout au long de l'étude avaient des valeurs inférieures à la limite de détection/quantification pour tous les sujets, indiquant l'absence d'exposition systémique significative des sujets au vecteur.
Pharmacocinétique clinique et excrétion
Les études sur la biodistribution et l'excrétion de vecteurs n'étaient qu'informatives mais ont indiqué une absence d'exposition systémique après application localisée et cutanée de bérémagène géperpavec.
Vyjuvek n'a aucun effet ou a un effet négligeable sur l'aptitude à conduire des véhicules ou à utiliser des machines.
Les données non-cliniques issues des études conventionnelles avec administration de doses uniques et répétées dans le cadre d'études toxicologiques n'ont pas révélé de risque particulier pour l'homme.
Aucune étude de toxicité pour le développement et la reproduction des animaux n'a été menée.
Aucune étude n'a été menée pour évaluer les effets du bérémagène géperpavec sur la cancérogénèse, la mutagenèse ou l'altération de la fertilité.
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient des organismes génétiquement modifiés (voir la rubrique Mises en garde spéciales et précautions d'emploi). Au cours de la préparation, de l'administration et de l'élimination, des précautions appropriées doivent être prises. Un équipement de protection individuelle (p. ex. des gants, un masque et une protection oculaire) doit être porté lors de la manipulation de Vyjuvek.
Les professionnelles de santé ou les aidantes enceintes ne doivent pas administrer Vyjuvek et ne doivent pas entrer en contact direct avec des plaies traitées, ni avec tous les matériaux qui ont été en contact avec des plaies traitées.
Préparation avant l'administration
Suivez les étapes ci-dessous pour la préparation de Vyjuvek.
Chaque carton contient un flacon de suspension (1 mL de volume extractible contenant 5×109 UFP) et un flacon de gel d'excipient (1,5 mL).
La concentration du médicament est de 2×109 UFP/mL après reconstitution.
Tableau 5. Étapes de préparation préalables à l'administration
|
Avant utilisation, les flacons congelés doivent être enlevés du carton et laissés à température ambiante. (Étape 1). Une fois les flacons décongelés (pendant environ 30 minutes), ils ne peuvent pas être recongelés. (Étape 2) Inspectez visuellement le flacon de suspension. La suspension peut contenir des particules de couleur blanche à blanc cassé qui sont inhérentes au produit. La couleur de la suspension peut varier de jaune opalescent à incolore. N'utilisez pas ce médicament si vous remarquez une décoloration. Inspectez visuellement le flacon de gel. Le gel est clair, incolore et visqueux. N'utilisez pas le gel si vous remarquez des particules ou une décoloration. Retournez doucement le flacon de suspension 4 à 5 fois pour mélanger le contenu. Retirez les opercules des flacons et nettoyez chaque bouchon du flacon à l'aide d'une compresse imbibée d'alcool. Laissez-les sécher. |
Étape 1 |
Étape 2 |
|
|
|
|
|
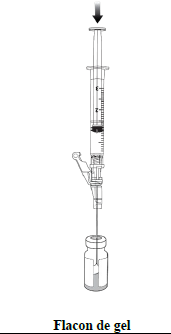
En utilisant une technique aseptique, prélevez 1 mL de suspension décongelée (Étape 1) à l'aide d'une seringue de 3 mL munie d'une aiguille (p. ex. 16G ou 18G). Transférez 1 mL de suspension décongelée dans le flacon de gel décongelé. (Étape 2). |
Étape 1 |
Étape 2 |
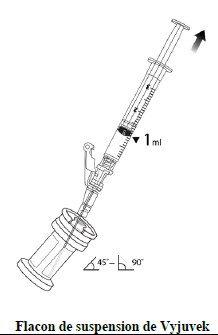 |
|
|
|
Sans retirer l'aiguille du flacon de gel, tirez l'aiguille afin qu'elle se trouve au-dessus du liquide, retirez 1 mL d'air (poche d'air) pour ventiler le flacon de gel après l'ajout de 1 mL de suspension de Vyjuvek, puis retirez la seringue et l'aiguille et jetez-les. Le flacon contenant la suspension et le gel combinés sera désigné sous le nom de flacon de Vyjuvek pour le reste des présentes instructions. |
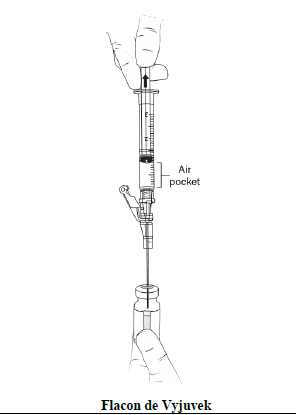 | |
|
Placez une compresse imbibée d'alcool sur le bouchon du flacon de gel et agitez le flacon vigoureusement à la main pendant au moins 10 secondes. Le gel d'excipient doit se mélanger à la suspension pour former un gel homogène. Inspectez visuellement le flacon de Vyjuvek. Le gel contenant la substance active peut contenir des particules de couleur blanche à blanc cassé qui sont inhérentes au produit. Le produit reconstitué, comme la suspension, peut varier en couleur du jaune opalescent à l'incolore. N'utilisez pas ce médicament si vous remarquez une décoloration. |
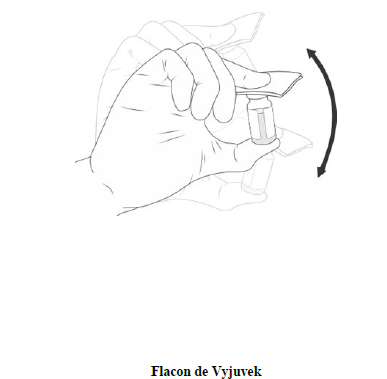 | |
|
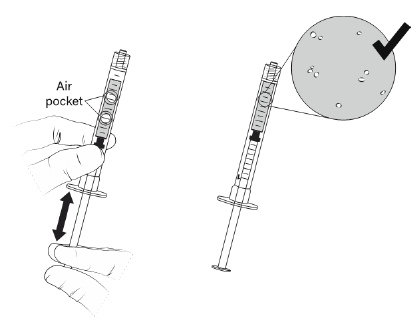
Raccordez une nouvelle aiguille (p. ex. 16G ou 18G) à une seringue de 1 mL et prélevez lentement 0,5 mL de Vyjuvek (Étape 1). Ne retournez pas le flacon pour retirer la seringue de Vyjuvek. Sans retirer l'aiguille du flacon, soulevez l'extrémité de l'aiguille au-dessus de la solution de Vyjuvek et débranchez la seringue, en laissant l'aiguille à l'intérieur du bouchon du flacon (Étape 2). Une poche d'air peut se former, ce qui est normal. |
Étape 1 |
Étape 2 |
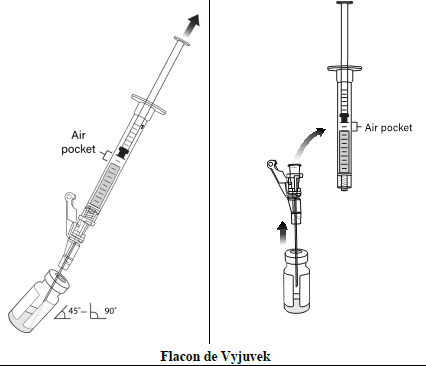 |
||
|
Manipulez doucement le piston vers le haut et vers le bas pour éliminer la poche d'air. NE TAPOTEZ PAS la seringue pour éliminer la poche d'air. Il peut subsister de petites bulles, ce qui est normal. |
| |
|
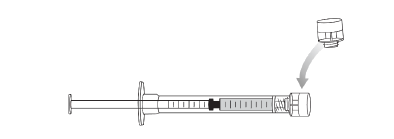
Refermez la seringue et laissez-la de côté. |
| |
|
Retirez l'autre seringue de 1 mL et reliezla à l'aiguille du bouchon du flacon de gel. Retirez 0,5 mL de Vyjuvek, retirez la poche d'air et bouchez la seringue. Le volume extractible est de 2,0 mL (4×109 UFP). Le cas échéant, répétez l'opération en fonction de la posologie recommandée. Étiquetez la seringue avec l'identifiant du patient, le nom du produit, le numéro de lot, la date de péremption et les conditions de conservation. Évitez de masquer les marques de la seringue nécessaires à l'application. Placez les seringues de Vyjuvek munies d'un bouchon dans un sachet en plastique étanche. Étiquetez le sachet en plastique avec l'identifiant du patient, le nom du produit, le numéro de lot, la date de péremption et les conditions de stockage. Il ne doit pas être utilisé plus de 2 mL (quatre seringues de 0,5 mL) au cours de la même semaine, car il s'agit de la dose hebdomadaire maximale.
|
| |
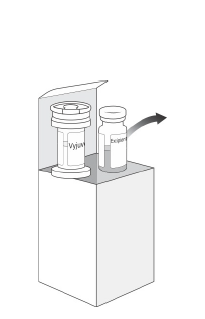


Placez le sachet en plastique étanche contenant des seringues de Vyjuvek dans un emballage tertiaire isotherme approprié («emballage extérieur») afin de maintenir une température de transport comprise entre 2 °C et 8 °C et de le protéger de la lumière.
L'emballage extérieur doit être entièrement fermé pour le transport.
L'emballage extérieur conçu pour le transport des seringues de Vyjuvek préparées, ne sera ouvert que sur le lieu d'administration.
Réception et stockage au lieu de l'administration
Après réception de l'emballage extérieur, conservez-le à température ambiante dans un endroit sécurisé, propre, hors de portée des enfants et exempt de toute contamination potentielle.
Seule la personne responsable de l'administration doit ouvrir l'emballage extérieur.
La personne responsable de l'administration doit vérifier que celui-ci est intact et qu'il n'y a aucun signe d'écoulement avant utilisation (voir la rubrique Posologie et mode d'administration).
Mesures à prendre en cas d'exposition accidentelle
En cas d'exposition accidentelle, il convient de suivre les recommandations locales relatives aux déchets pharmaceutiques.
Toutes les surfaces susceptibles d'avoir été en contact avec du bérémagène géperpavec doivent être nettoyées et tous les écoulements doivent être désinfectés avec un agent virucide tel que l'alcool isopropylique à 70 %, le peroxyde d'hydrogène à 6 % ou le chlorure d'ammonium à moins de 0,4 %.
En cas d'exposition accidentelle par éclaboussures des yeux ou des muqueuses, rincez à l'eau pendant au moins 5 minutes.
En cas de contact avec une peau intacte ou en cas de lésion par piqûre d'aiguille, nettoyez soigneusement la zone touchée à l'aide de savon et d'eau et/ou d'un désinfectant.
Précautions à prendre pour l'élimination du médicament
Tout médicament non utilisé ou tout déchet matériel (p. ex. flacon, seringue, aiguille, matériel de nettoyage) susceptible d'avoir été en contact avec Vyjuvek doit être éliminé conformément aux recommandations locales en matière de déchets pharmaceutiques.
Désinfectez les pansements avec un agent virucide, tel que l'alcool isopropylique à 70 %, le peroxyde d'hydrogène à 6 % ou le chlorure d'ammonium à moins de 0,4 %, et éliminez les pansements désinfectés dans un sachet en plastique étanche distinct avec les déchets ménagers ou conformément aux exigences locales.
Les premières administrations doivent être effectuées en milieu hospitalier.
Liste I.
Prescription hospitalière.
Prescription réservée aux spécialistes et services DERMATOLOGIE.
Suspension et gel pour gel.
La suspension est de couleur jaune opalescent à incolore après décongélation, depuis l'état congelé.
Le gel est visqueux et clair après décongélation.
Chaque carton de Vyjuvek contient un flacon de suspension et un flacon de gel.
Suspension
Volume extractible de 1 mL contenant 5×109 UFP dans un flacon en copolymère de cyclo-oléfines muni d'un bouchon en élastomère thermoplastique et d'un opercule vert.
Gel
Volume de remplissage de 1,5 mL dans un flacon en verre de type 1 muni d'un bouchon en élastomère de bromobutyle et d'un opercule bleu.
Bérémagène géperpavec est un vecteur de thérapie génique dérivé du virus Herpes simplex de type 1 HSV-1, incompétent pour la réplication, qui a été génétiquement modifié pour exprimer le collagène humain de type VII (COL7) sous le contrôle du promoteur du cytomégalovirus humain (hCMV).
Bérémagène géperpavec est produit dans les cellules Vero par la technologie de l'ADN recombinant.
Chaque flacon contient 1 mL de volume extractible de suspension contenant 5×109 unités formant plage (UFP) de bérémagène géperpavec.
Après avoir mélangé 1 mL de la suspension avec le gel, Vyjuvek contient 5×109 UFP dans 2,5 mL. Le volume extractible est de 2,0 mL (4×109 UFP).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Suspension
Glycérol (E422)
Chlorure de sodium
Phosphate dissodique (E339)
Chlorure de potassium (E508)
Hydrogéno-orthophosphate dipotassique (E340)
Gel
Hypromellose (E464)
Trométamol
Chlorure de sodium
Phosphate dissodique (E339)
Hydrogéno-orthophosphate dipotassique (E340)